
https://erowid.org/plants/kratom/kratom_journal2.shtml
Sections
TABLES 2-5 Alkaloids isolated from species of Mitragyna
Summary
TABLES 6 AND 7 Table 6: Open E ring alkaloids Alkaloids present in Mitragyna speciosa Korth
Table 7: Closed E ring alkaloids
Alkaloids present in Mitragyna javanica Koord and Valeton TABLE 8
Alkaloids isolated from Mitragyna javanica
Acknowledgements
Details
Author: E.J. SHELLARD
Pages: 41 to 55
Creation Date: 1974/01/01
The alkaloids of Mitragyna with special reference to those of Mitragyna speciosa, Korth
Professor of Pharmacognosy E.J. SHELLARD
Pharmacognosy Research Laboratories, Department of Pharmacy, Chelsea College, University of London
The genus Mitragynabelongs to the family Rubiaceae and is found in swampy territory in the tropical and sub-tropical regions of Africa and Asia.
Mainly arborial in character, some species growing to a height of 30 metres, Mitragynaspecies are characterized by the globular flowering head each containing up to 120 florets. Each floret is surrounded by many overlapping bracteoles which completely cover the developing florets during the flower bud stage. The inflorescence is a dichasial cyme. The fruit is a capsule containing numerous small flat seeds. The young woody shoots bear 10-12 leaves arranged in opposite and decussate pairs each pair of leaves being accompanied by two interpetiolar stipules which initially are closely appressed and protect the apical bud.
The genus was given the name Mitragynaby Korthals because the shape of the stigmas in the species he examined resembled a bishop’s mitre. However the nomenclature has frequently been confused, the genus being variously named as Nauclea, Sarcocephalus, Stephegyne and Uncaria though consistently recognised as members of the tube Naucleeae in the sub-family Naucleoideae. There has also been some confusion at species level but today the species are recognized as follows:
West Africa
Mitragyna inermis (Willd.) O. Kuntze ( M. africana (Willd.) O. Kuntze)
Mitragyna ciliata Aubrev. and Pellegr. ( M. macrophylla Hiern)
Mitragyna stipulosa (D.C.) O. Kuntze ( M. macrophylla Hiern)
East Africa
Mitragyna rubrostipulata Havil.
India and S.E. Asia
Mitragyna hirtusa Havil.
Mitragyna javanica Koord. and Valeton.
Mitragyna parvifolia (Roxb.) Korth.
Mitragyna rotundifolia (Roxb.) O. Kuntze (N. diversifolia (Hook.f.) Havil.)
Mitragyna speciosa Korth.
Mitragyna tubulosa Havil.
Mitragyna brunnonsis (Wall ex G.Don) has been included in M. rotundifolia.
In a more recent revision of the genus (1972) R.C. Bakhuisen van den Brink (Leiden Herbarium) has included Mitragyna javanica in M. parvifolia though whether this can be justified on chemotaxonomic grounds remains to be argued.
In 1897 Ridley reported the leaves and bark of Mitragyna speciosa as a cure for the opium habit and this was quoted by Hooper (1907) In 1907 Holmes had referred to the leaves and possibly, the leaves of M. parvifolia as well, as an opium substitute. Certainly the leaves of M. speciosa have been chewed for many years under the local name ‘Kratom’ by the native population of Thailand as a stimulant though the practice is now forbidden. As a consequence the leaves of M. javanica are frequently used as a substitute but are not considered to be as effective. The natives will also distinguish between different Kratoms, for example, those with red and those with green midribs (Tantivatana, 1965).
Hooper actually isolated an alkaloid from the leaves of M. speciosa and this was repeated in 1921 by Field who named the alkaloid mitragynine. Following this quite an amount of work was carried out on species of Mitragyna from Afr
ica and South East Asia particularly by Raymond-Hamet. By 1940 three alkaloids in addition to mitragynine had definitely been characterized viz. mitraphylline from the bark of M. rubrostipulata (Michiels and Leroux 1925), rhynchophylline from the bark of M. stipulosa (Larrieu, 1930) and rotundifoline (Barger, Dyer and Sargent, 1939). Raymond-Hamet isolated an alkaloid from M. inermis which he named mitranermine (1934) while Denis named alkaloids he obtained from M . speciosa mitraversine and mitraspecine (1937) but these were probably mixtures of alkaloids.
Since mitragynine was the only constituent isolated from Mitragyna speciosa it was assumed to be the physiologically active constituent having morphine-like properties, Grewel (1932) reported to be a protozoal poison but in 1933 Raymond-Hamet and Millat undertook a more critical examination and reported it to have markedly depressant properties. This was substantiated in 1934 by Masson. More recently Macko, Weisbach and Douglas (1972) reported that mitragynine possesses pain threshold elevating and antitussive properties comparable with those of codeine but with no emesis. There were no addictive properties as may be found in morphine.
In the post-war period 1950-60 further work was undertaken on the genus but only one new alkaloid was reported, isorhynchophylline from M. rubrostipulata (Seaton, Tondeur and Marion, 1958) and this was considered to be an isomer of the alkaloid previously isolated by Larrieu.
None of these workers, however, had the advantages of the modern techniques of separation and characterization and since the emphasis upon research in the Pharmacy Department, Chelsea College had until this time been concerned with synthetic substitutes for morphine it was decided to re-investigate the genus for alkaloids.
To date 40 alkaloids have been isolated and characterized. These are given in tables 2-5 (pp. 47-48).
Although mitragynine was the first alkaloid to be isolated (Hooper, 1907) its structure was not finally determined until 1964 when Zacharias, Rosenstein and Jeffrey showed by X-ray crystallography that the C(17)-H is cis to the methyl ester at C(16). Many workers had previously been involved with the determination of its general structure and it was shown by Joshi, Raymond-Hamet and Taylor (1963) to be an indole having a methoxy group in the C(19) position and an open E ring (E seco).
The first Mitragyna alkaloid to have its structure determined was mitraphylline. This was achieved in 1958 by Seaton, Tondeur and Marion who showed it to be an oxindole without substitution in the C(9) position and having a closed E ring. In 1960 Seaton, Nair, Edwards and Marion converted mitraphylline to its isomer-isomitraphylline. Wenkert, Wickberg and Leicht (1961) suggested that the isomerization occurred at the spiro C(7).
The naturally occurring isomitraphylline was first obtained in 1966 by Shellard and Philtipson who isolated it from the leaves of M. speciosa together with a further isomer which they named speciophylline.
Following extensive work by many people, Seaton, Nair, Edwards and Marion, in 1960, proposed structures for rhynchophylline and isorhynchophylline as oxindole alkaloids without substitution in the C(9) position and having the E seco structure.
With the isolation of the well known alkaloid ajmalicine from the leaves of M. speciosa (Beckett, Shellard, Phillipson and Lee, 1966) it was established that the Mitragynine alkaloids were either indole or oxindole alkaloids having a closed or open E ring with substitution occurring in some alkaloids in the C(9) position.
The structure of rotundifoline first isolated by Barger, Dyer and Sargent in 1939 presented many problems and it was not until an isomer isorotundifoline was isolated almost simultaneously from M. stipulosa (Beckett, Shellard and Tackie, 1965) and M. parvifolia (Shellard and Phillipson, 1964) that it was possible for Beckett and Tackie to propose its real structure as an E seco oxindole with C(9) – OH. The striking difference between these two isomers arising from the isomerism about C(7) is the hydrogen bonding which occurs between the C(9) – OH and the N(4) in rotundifoline but not in isorotundifoline, so that the former does not possess phenolic properties.
FIGURE 1 Structure of the Mitragyna alkaloids
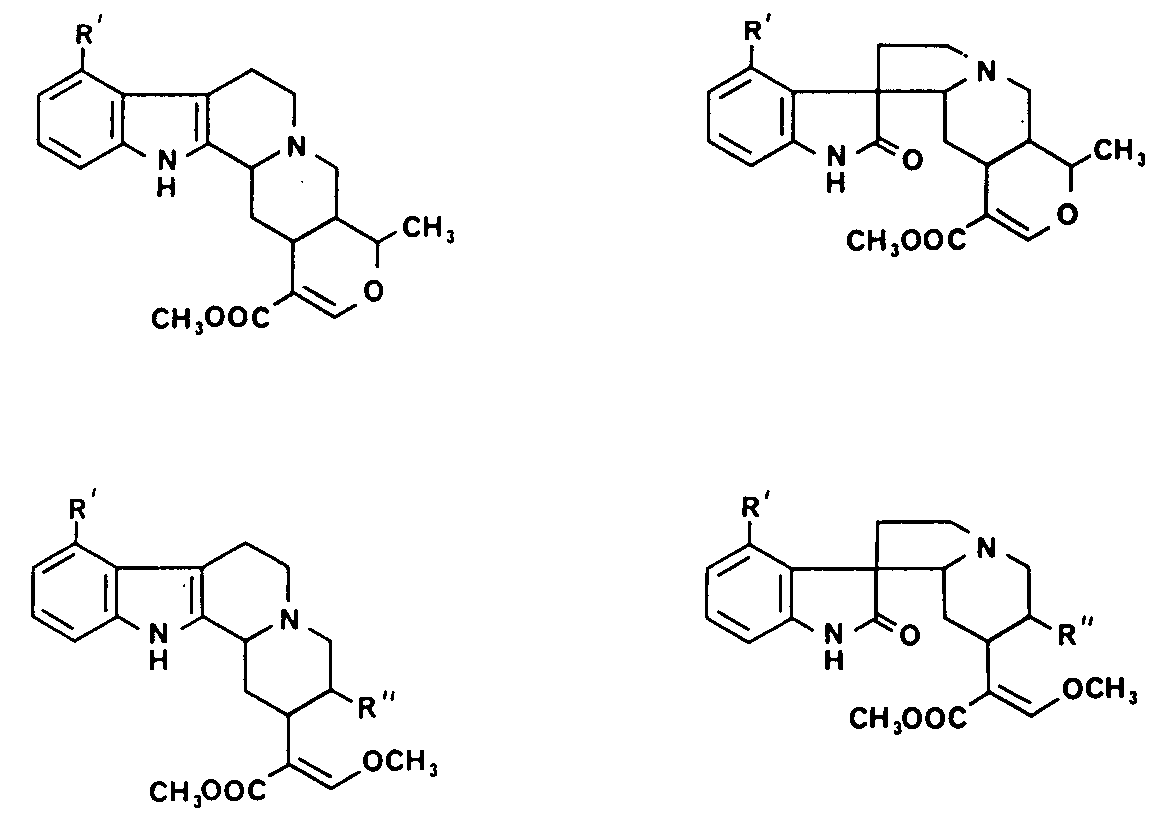 Figure 1 shows the different types of alkaloid in Mitragynine species. It will be seen that all the alkaloids have asymmetric centres at C(3), C(15) and C(20) and that the closed E ring alkaloids also have asymmetric centre at C(19). The E seco alkaloids may show geometric isomerization because of the double bond between C(16) and C(17) though all the alkaloids of known absolute configuration possess a C(17) – H cis to the ester group at C(16). In addition the oxindole alkaloids have an asymmetric centre at C(7), those alkaloids in which the lactam carbonyl lies below the plane of the C ring being termed the A series and those in which the lactam carbonyl lies above the plane of the C ring being termed the B series. Further, in both types of oxindole alkaloids the lone pair of electrons on N(4) may either be on the same side of the C(7) as the lactam carbonyl group or on the opposite side; the former are known as syn and the latter as antialkaloids. The oxindole alkaloids readily isomerize about C(7) and C(3) to give a mixture of the isomers, except that the pseudo oxindoles are not considered to be stable.
Figure 1 shows the different types of alkaloid in Mitragynine species. It will be seen that all the alkaloids have asymmetric centres at C(3), C(15) and C(20) and that the closed E ring alkaloids also have asymmetric centre at C(19). The E seco alkaloids may show geometric isomerization because of the double bond between C(16) and C(17) though all the alkaloids of known absolute configuration possess a C(17) – H cis to the ester group at C(16). In addition the oxindole alkaloids have an asymmetric centre at C(7), those alkaloids in which the lactam carbonyl lies below the plane of the C ring being termed the A series and those in which the lactam carbonyl lies above the plane of the C ring being termed the B series. Further, in both types of oxindole alkaloids the lone pair of electrons on N(4) may either be on the same side of the C(7) as the lactam carbonyl group or on the opposite side; the former are known as syn and the latter as antialkaloids. The oxindole alkaloids readily isomerize about C(7) and C(3) to give a mixture of the isomers, except that the pseudo oxindoles are not considered to be stable.
TABLE 1
Configuration of the Mitragyna alkaloids
| C(3) | C(20) | |
|---|---|---|
| ALLO | ? | ? |
| NORMAL | ? | ? |
| EPIALLO | ? | ? |
| PSEUDO | ? | ? |
FIGURE 2 Preferred conformations of corynantheidine-type alkaloids
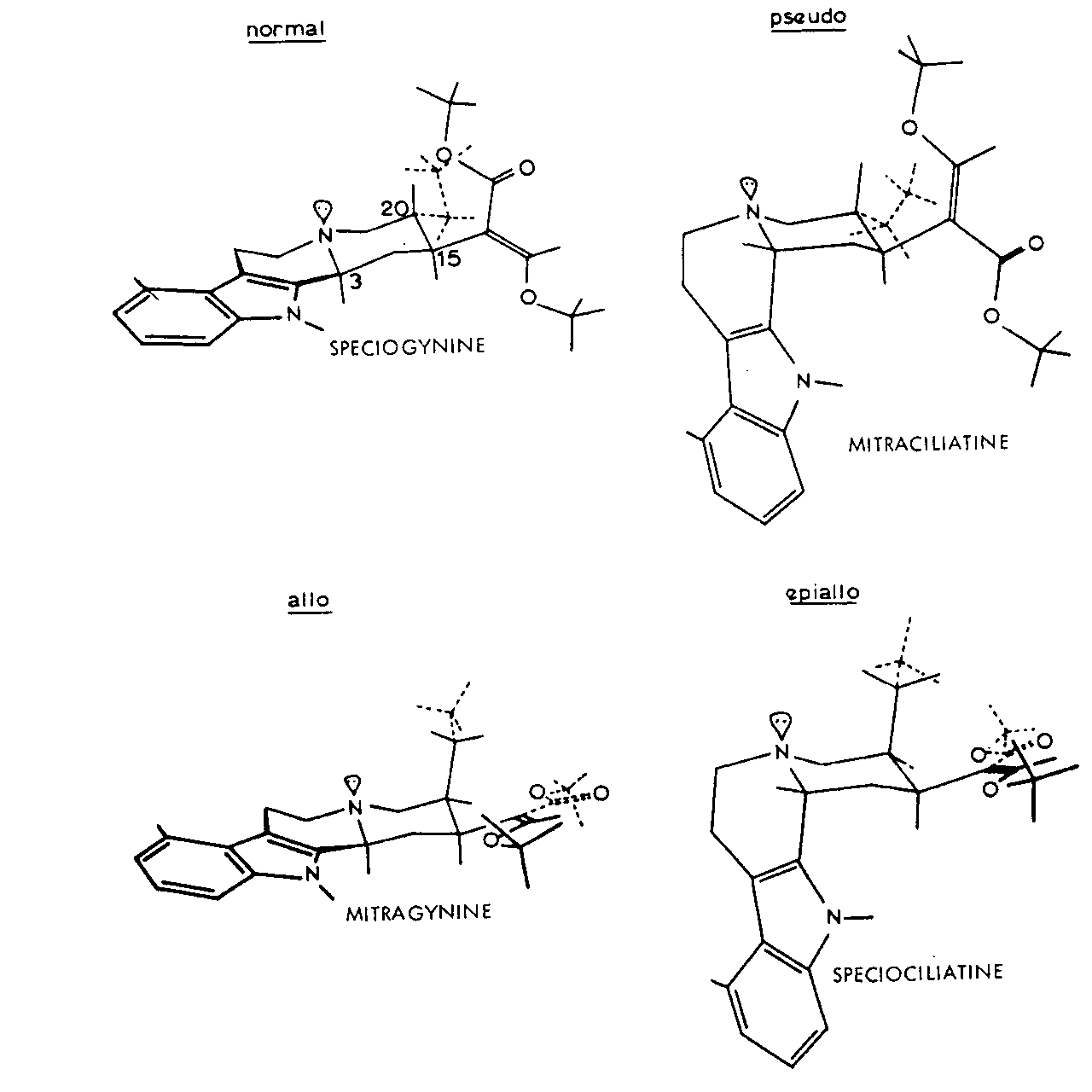 The possible configuration and nature of the alkaloids are shown in table 1. All have C(15)-H? and C(19) – H? so that only C(3) – H and C(20) – H are involved. Substitution may occur in the available ring at C(9) and is either a methoxy or hydroxy group. In the E seco alkaloids R” may be CH 2CH 3or CH=CH 2.
The possible configuration and nature of the alkaloids are shown in table 1. All have C(15)-H? and C(19) – H? so that only C(3) – H and C(20) – H are involved. Substitution may occur in the available ring at C(9) and is either a methoxy or hydroxy group. In the E seco alkaloids R” may be CH 2CH 3or CH=CH 2.
Work by Trager, Lee, Phillipson and others at Chelsea around 1967-8 has made it possible to elucidate the configuration and conformation of all the Mitragyna alkaloids. The preferred conformations of the Open E ring alkaloids are shown in figure 2.
When alkaloids present in individual species are considered in terms of their configuration and D/E ring structure, if both indole and oxindole alkaloids are present the D/E ring systems are identical. Furthermore with the exception of M. speciosa the indole alkaloids present in the largest quantities are those with the thermodynamically least stable configuration, i.e. epiallo and pseudo. This gave rise to the postulation that there could be a well defined biogenetic link between the formation of the indole and the oxindole alkaloids and bearing in mind that vincoside, one of the indole alkaloid precursors was considered to have a C(3) – H? it was suggested that the plants synthesize the thermodynamically more stable indole alkaloids which then isomerize, and that all the indoles then give rise to the corresponding oxindoles (Shellard, Phillipson and Gupta, 1970).
This is summarized as-
?allo indoles? allo oxindoles A and B
epiallo indoles? epiallooxindoles A and B
?normal indoles? normal oxindoles A and B
pseudo indoles
The indole transformation involves the conversion of C(3)H-? to C(3)H-? but this was achieved by in vitro by Wenkert and Roychoudhury (1956, 1957). The in vitro conversion of indole alkaloids to the corresponding oxindoles was carried out by Finch and Taylor (1962) and Shavel and Zinnes (1962). Since then these conversions have been repeated with most of the Mitragyna indole alkaloids.
Such a relationship certainly appears to occur among the alkaloids present in many of the plants; for example, in the leaves of Mitragyna parvifolia from Burma the alkaloids present are:
?dihydrocorynantheine? isorhynchophylline ? rhynchophylline
hirsutine
while in those from the Maharashtra State of India and from Ceylon the alkaloidal sequence is: isopteropodine
?tetrahydroalstonine?speciophylline ?pteropodine
However, in some of the plants examined, the allo and normal indole alkaloids could not be detected. Nevertheless, our speculative hypothesis required them to be present even if only in small quantities so we examined leaves, stem bark and roots collected at monthly intervals from the same trees over a large period of time. The alkaloids were found in trace amounts in the very young leaves. With Mitragyna species there is no seasonal leaf-fall; after some time the leaves fall and a short time afterwards new leaves appear.
However, in one of the geographical variants of Mitragyna parvifolia although ajmalicine could not be found, 3-isoajmalicine is present and the thought that in this species, mitraphylline could be obtained from 3-isoajmalicine was shown to be correct by in vitro and in vivo experiments (Shellard and Sarpong, 1971, Shellard and Houghton, 1972). Other feeding experiments showed that mitraphylline is obtained from ajmalicine but the unexpected observation was that there is no interconversion between the two related indole alkaloids. Later work using 14C-labelled indole alkaloids also showed that there is no interconversion between the allo and epiallo indole alkaloids though each type of alkaloid would give rise to both allo and epiallo oxindole alkaloids.
It was therefore considered more likely that the C(3)-H ?and C(3)-H indole alkaloids were separately synthesized from a precursor but that the epiallo and pseudo indole alkaloids were the dominant ones and offered the main pathway to the oxindole alkaloids. This modification of our hypothesis later became more rational with Black-stock’s revelation in 1972 that the C(3)-H in vincoside was actually ?. Following further in vivo experiments using both labelled and unlabelled alkaloids the hypothesis has been modified to meet these newly observed facts and is now represented as follows- indoles oxidoles A and B

C(3)H? C(3)H? major route minor routes
It might be mentioned, in passing, that the existence of mitraphylline in some species cannot be explained on the basis of the hypothesis and it has been shown by Shellard and Houghton (1973) following feeding experiments, that there is an interconversion between rhynchophylline and mitraphylline via the intermediary corynoxeine (20 vinyl rhynchophylline). Using the method of Djakoure et al. (1972) this conversion has now been achieved in vitro.
An interesting feature and one which may be of special significance is that at no time during the feeding experiments were any changes observed in the C(9) group of the indole alkaloids which indicates that methoxylation and demethoxylation did not take place and suggests that the enzyme systems involved are not present in the plants. Such processes must however occur among the oxindoles since there is no evidence of C(9)-OH indole alkaloids to correspond to the C(9)-OH oxindoles which are present.
Thus the indole-oxindole combinations in Mitragyna can be divided into 8 separate groups. The names of the alkaloids and their position in these sequences are given in tables 2-5.
TABLES 2-5 Alkaloids isolated from species of Mitragyna
Table 2: Open E ring alkaloids
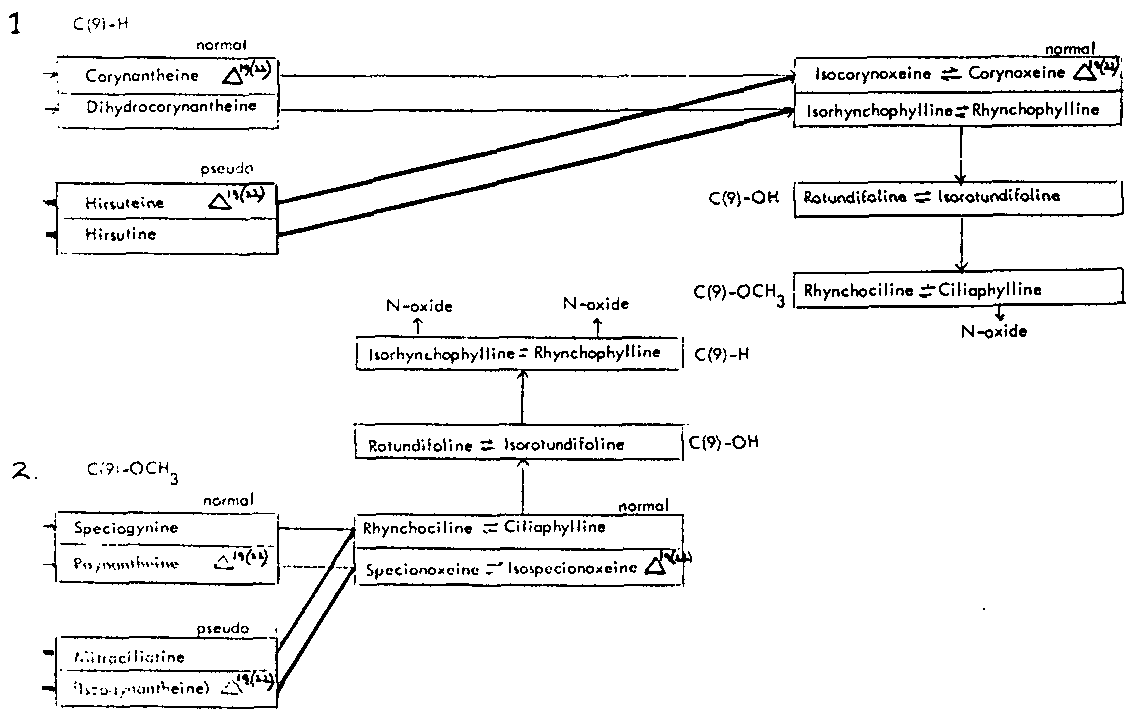
( ) semisynthetic, i.e. prepared from naturally occurring Mitragyna alkaloids.
Table 3: Open E ring alkaloids
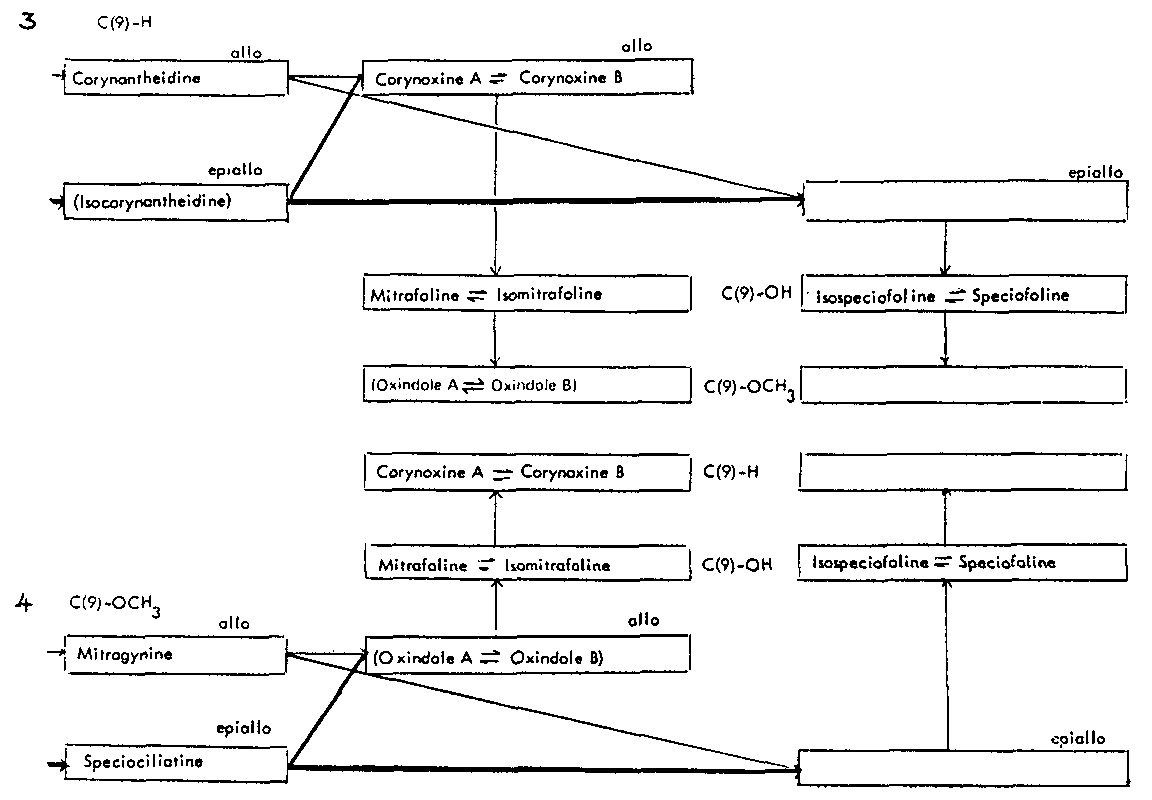 ( ) semisynthetic, i.e. prepared from naturally occurring Mitragyna alkaloids.
( ) semisynthetic, i.e. prepared from naturally occurring Mitragyna alkaloids.
Table 4: Closed E ring alkaloids
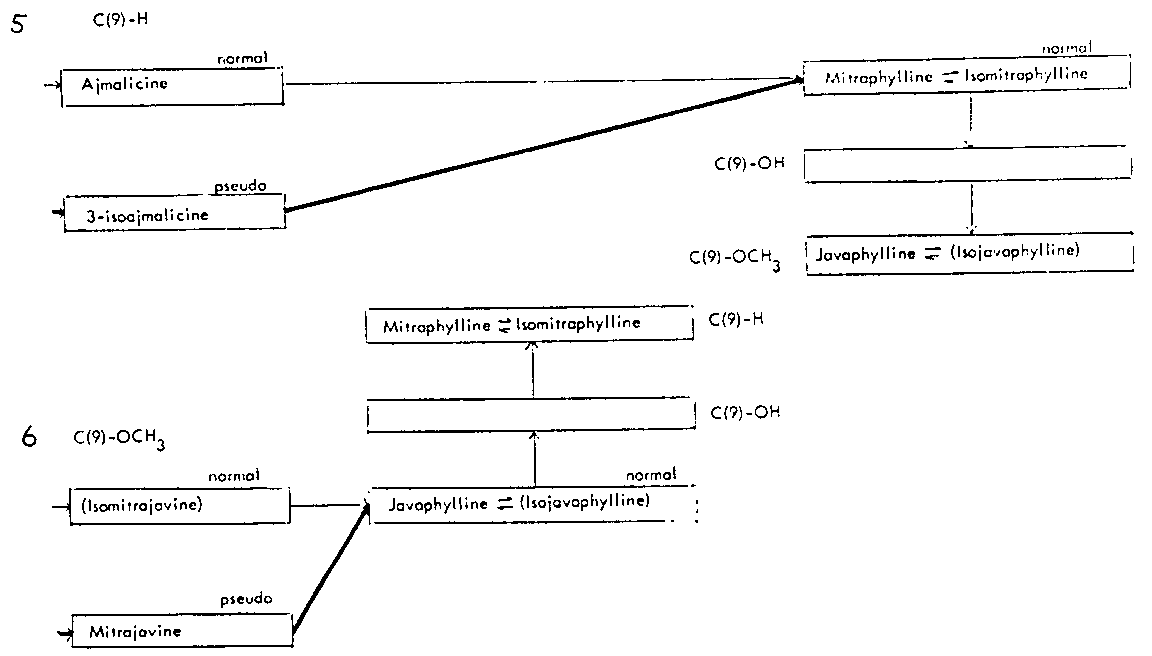 ( ) semisynthetic, i.e. prepared from naturally occurring Mitragyna alkaloids.
( ) semisynthetic, i.e. prepared from naturally occurring Mitragyna alkaloids.
Table 5: Closed E ring alkaloids
 It is within this general context of the Mitragyna alkaloids that consideration is now given to those isolated from the leaves of Mitragyna speciosa in the Pharmacognosy Research Laboratories at Chelsea College. Altogether 30 samples of leaves have been examined and the results are given below-
It is within this general context of the Mitragyna alkaloids that consideration is now given to those isolated from the leaves of Mitragyna speciosa in the Pharmacognosy Research Laboratories at Chelsea College. Altogether 30 samples of leaves have been examined and the results are given below-
| Date of collection | Origin | Alkaloids isolated | Other alkaloids present |
|---|---|---|---|
| 14. October 1963 | Thailand | as No. 4 | |
| (Kratom) | |||
| 15. December 1963 | Thailand | as No. 4 | |
| (Kratom) | |||
| 16. 1965 | New Guinea | mitragynine, | not known |
| speciogynine, | |||
| speciociliatine, | |||
| paynantheine, | |||
| specionoxeine, | |||
| isospecionoxeine | |||
| 17. April 1967 | Thailand | as No. 10 | |
| plus two oxindoles | |||
| (epiallo ?) | |||
| 18-30 12 monthly | Thailand | mitragynine, | |
| samples | speciogynine, | ||
| collected | speciociliatine, | ||
| from 1969-70 | mitraciliatine, | (traces in some months) | |
| paynantheine, | |||
| ajmalicine, | (traces in some months) | ||
| corynoxine A and B, | |||
| mitrafoline, | |||
| isomitrafoline, | |||
| isospeciofoline, | |||
| speciofoline, | |||
| The A and B oxindole isomers corresponding to | |||
| isocorynantheidine and to mitragynine | |||
| (traces in some months) |
Summary
Altogether 22 alkaloids have been isolated from the leaves of Mitragyna speciosa but it will be seen that the alkaloidal content varies from location to location and from time to time. With regard to the Thailand material there appears to be some variation based upon different geographic origins (though unfortunately the precise places of collection are unknown) but within each geographical region there is a quantitative variation from month to month which transforms itself into a qualitative variation, certainly as far as the oxindole alkaloids are concerned. The main indole alkaloidal content is fairly stable and it would appear that mitragynine, speciogynine, paynantheine with small amounts of speciociliatine are present in all leaves. Mitragynine is the dominant alkaloid and is exclusive to Mitragyna speciosa. Paynantheine, the C(20)-CH=CH 2 counterpart to speciogynine and speciociliatine also appears to be specific to this species. Speciogynine and mitraciliatine on the other hand have been isolated from Mitragyna inermis.
The oxindole content shows tremendous variation, both from location to location and from time to time and usually occurs in small or trace amounts when present. It is interesting to note that the first oxindole alkaloid isolated from this species was a C(9)-OH alkaloid which was isomeric with rotundifoline and isorotundifoline (Beckett, Shellard and Tackie, 1965) but could not at that time be fully characterized.
TABLES 6 AND 7 Table 6: Open E ring alkaloids Alkaloids present in Mitragyna speciosa Korth
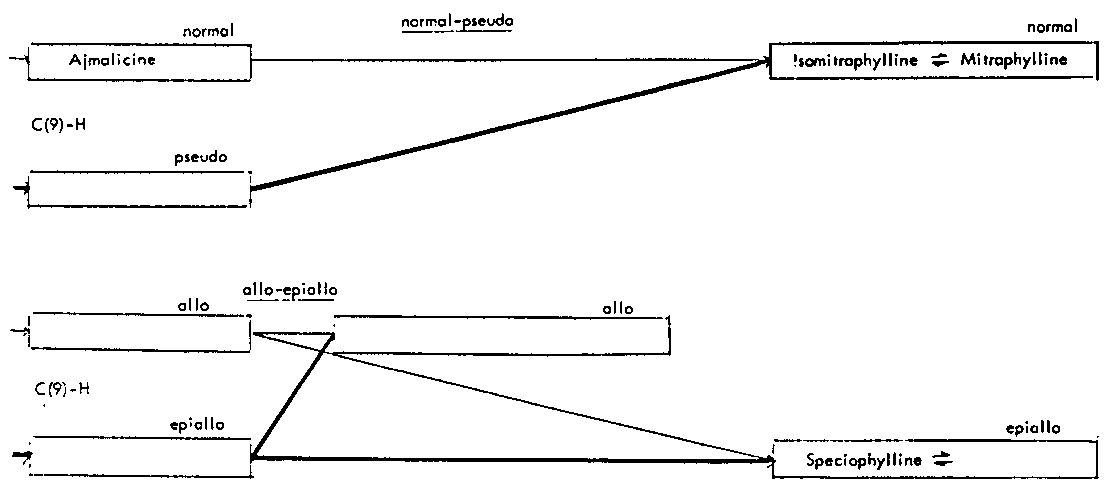
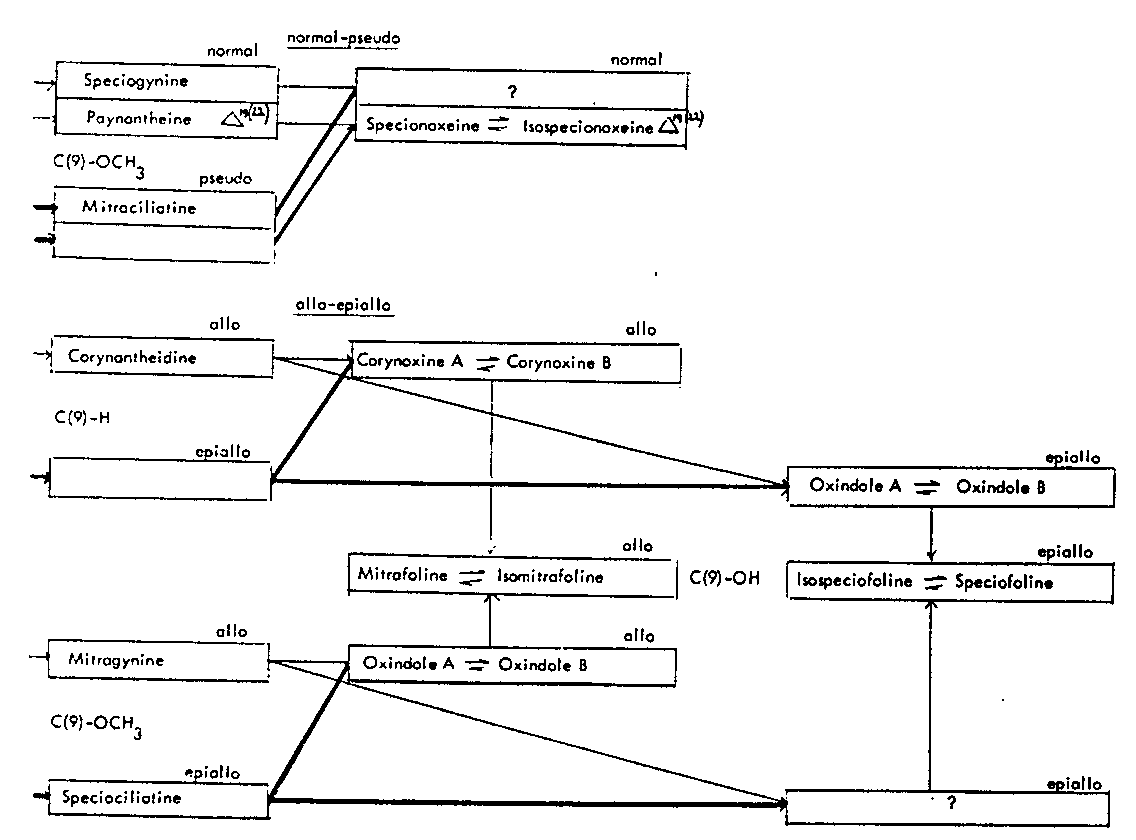
Table 7: Closed E ring alkaloids
It has now been isolated again in larger quantities (Houghton and Shellard, 1974) though from material collected in Thailand in 1962 and 1963 as well as more recently. These leaves also contained its isomer–isospeciofoline and the two alkaloids have now been characterized as epiallo C(9)–OH open E ring oxindole alkaloids (Hemingway, Houghton, Phillipson and Shellard, 1974). Some of the plant material also contained two other C(9)–OH open E ring oxindole alkaloids (Houghton and Shellard, 1974) and these were shown to be the allo isomers (Hemingway, Houghton, Phillipson and Shellard, 1974). It is worth noting that a different sample of leaf, obviously collected from a different source, but provided at the same time, had no evidence of these alkaloids. The alkaloids have been named mitrafoline and isomitrafoline. The leaves collected on December 1962 (a) and April 1963 (a) and later in April 1967 also contained the C(9)-H allo open E ring oxindole alkaloids, corynoxine A and B which had not previously been isolated from a species of Mitragyna. There is some evidence that the leaves collected in April 1967 also contain the corresponding C(9)-H epiallo open E ring alkaloids. These alkaloids-corresponding to the indole alkaloids isocoryn-antheidine (which has not yet been obtained from natural sources) also occur in some of the leaves collected at regular monthly intervals from the same tree growing near the University in Bangkok as also do the C(9)-OCH 3 allo open E ring oxindole alkaloids corresponding to the indole alkaloid mitragynine. In some months there are appreciable quantities of these hitherto undescribed oxindole alkaloids. The leaves also contain varying amounts of corynoxine A and B, mitrafoline and isomitrafoline, isospeciofoline and speciofoline.
It was in some of these leaves that small quantities of mitraciliatine were detected, this being the first occasion the alkaloid so closely related to the mitragynine group had been observed in Mitragyna speciosa. As expected the four main indole alkaloids occurred every month though the quantities present varied and in some months traces of corynantheidine and ajmalicine were present.
One oxindole alkaloid present only in some leaves and first isolated from Mitragyna speciosa is speciophylline (Beckett, Shellard, Phillipson and Lee, 1966). Speciophylline is the epiallo A isomer corresponding to the normal closed E ring oxindole alkaloids, mitraphylline and isomitraphylline. These alkaloids are present in some leaves but not others-usually the three closed E ring C(9) – H oxindoles occur together. An interesting point however, is that the corresponding epiallo B isomer-uncarine F – has never been detected in Mitragyna speciosa.
Reference must be made to the plant material from New Guinea which contained two oxindole alkaloids having C(20) – CH=CH 2 groups and which were normal open E ring alkaloids, corresponding, therefore to the indole alkaloid paynantheine. These alkaloids were named specionoxeine and isospecionoxeine (Trager, Lee, Phillipson and Beckett, 1967). Unfortunately this material was not subjected to a complete examination so that no definite statement can be made about the total alkaloidal content.
If the alkaloidal sequences (based upon the Shellard, Houghton modified hypothesis 1973) are considered it will be seen that of the 7 sequences within the genus Mitragyna, 5 are represented in this one species though none of them are complete. It must be emphasized, however, that these alkaloids have all been isolated from the leaves only and that examination of the root bark may reveal the presence of alkaloids which would complete the sequences. Further, there is T.L.C. evidence of trace quantities of C(9) – OCH 3 oxindole alkaloids although until they can be isolated in sufficient quantities for characterization purposes it is not possible to indicate the sequence to which they belong (shown by ? in the tables).
The hypothesis referred to above and discussed earlier in the text suggests that the main route of biogenesis of the Mitragyna alkaloids is via the C(3) – H? and this is certainly substantiated by the alkaloids present in other species of Mitragyna. However, in Mitragyna speciosa the C(3) – H? alkaloids predominate, the only C(3) – H? indole alkaloids being speciociliatine which occurs in small amounts and mitraciliatine which was detected only in trace amounts in some of the leaves collected on a regular monthly basis. Corynantheidine and ajmalicine even when present in appreciable amounts do not have their corresponding C(3) – H? alkaloids present. Thus it would appear that the enzyme controlling the C(3) – H? pathway is dominant and as such would characterise this particular species.
The alkaloids of Mitragyna 53
The allo-epiallo open E ring sequence is almost complete and if the C(9) -H and C(9) -OCH 3 oxindoles as yet not identified completely should prove to fit into this sequence it is reasonable to suppose that the epiallo C(9) -H indole alkaloid (isocorynantheidine) would be present. So far this alkaloid has not been found to occur naturally. The normal-pseudo open E ring sequence contains no C(9) -H alkaloids and in the C(9) -OCH 3 sequence there is a possibility that the oxindole corresponding to speciogynine might be present. However there is no evidence to suggest that isopaynantheine might also occur.
Among the closed E ring sequences the absence of 3 isoajmalicine can be explained in view of the emphasis on this species of the C(3) -H? biogenetic route. There is, however, no valid explanation for the occurrence in some plants of speciophylline without its corresponding B isomer and in the absence of either tetrahydroalstonine or akuammigine and the allo oxindoles of the sequence.
Since it has been reported that the leaves of Mitragyna javanica are sometimes chewed as a substitute for those of Mitragyna speciosa it will be interesting to note that the sample of leaves of M. javanica obtained from Thailand in 1965 contained the alkaloids of the C(9) -H closed E ring normal-pseudo sequence and some alkaloids in the C(9) -OCH 3 closed E ring normal-pseudo sequence. (Shellard, Beckett, Tanti-vatana, Phillipson and Lee, 1967).
Alkaloids present in Mitragyna javanica Koord and Valeton TABLE 8
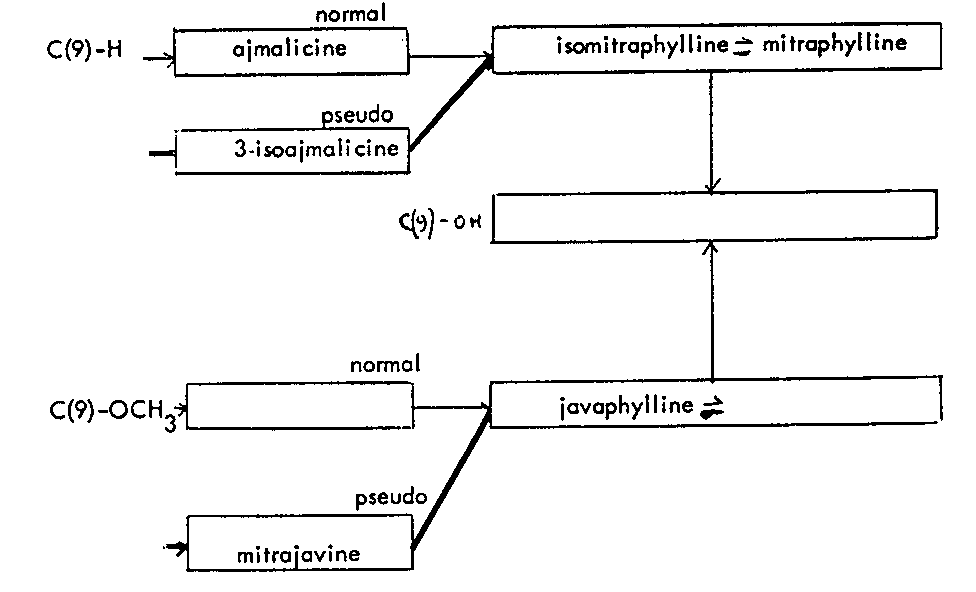
Alkaloids isolated from Mitragyna javanica
Mitrajavine was the dominant alkaloid but there was no evidence of the isomer, isomitrajavine while only the A isomer of the corresponding oxindole series was present. 3-isoajmalicine was present in larger amounts than ajmalicine and thus it appears that the route of biogenesis is through the C(3)-H? pathway. No pharmacological tests have been undertaken on mitrajavine.
Acknowledgements
The author would like to thank all his co-workers in the investigation of Mitragyna species and their names occur in the list of references. However he would like to acknowledge in particular the help of his former colleague, Dr. J. D. Phillipson (now Senior Lecturer in Pharmacognosy, School of Pharmacy (University of London), London, W.C.1.) and Dr. P. J. Houghton (now Lecturer in Pharmacognosy, Department of Pharmacy, Chelsea College (University of London) S.W.3.), for their special contribution towards the isolation and characterization of alkaloids from Mitragyna speciosa.
References
001Bakhuisen van den Brink R.C. private communication (1972).
002Barger, G., Dyer, E. and Sargent, K. J. J. Org. Chem., 4, 418 (1939).
003Beckett, A. H., Shellard, E.J., Phillipson, J. D. and Lee, C.M. Planta Medica, 14, 266 and 277 (1966).
004Beckett, A. H., Shellard, E.J. and Tackie, A.N. Planta Medica, 13, 241 (1965).
005Beckett, A.H. and Tackie, A.N.. Chem. and Ind. (Rev.) 1122 (1964).
006Blackstock, W. P., Brown, A. T. and Lee, G. K. Chem. Comm. 910 (1971).
007Denis P., Bull. Acad. Belg. Sci., 23, 174 (1937).
008Djakour?, L., Jarreau, F.X., Goutarel, R. and Janot, M.M. Compt. rend. Acad. Sci. Paris, 274, 1529 (1972).
009Field, E. J. Chem. Soc., 119, 887 (1921).
010Finch, N. and Taylor, N. I., J. Amer. Chem. Soc. 87 1318 and 3871 (1962).
011Grewel, K. S. J. Pharmacol. exp. Ther., 46, 273 (1932).
012Grewel, K. S. Brit. J. Med. Psychol., 12, 41, (1932).
013Hemmingway, S., Houghton, P. J., Phillipson, J. D. and Shellard, E. J. Phytochemistry (in the press) (1974)
014Holmes, E. M. J. Pharm. 78, 453 (1907).
015Hooper, D. J. Pharm., 78, 453 (1907).
016Joshi, B. S., Raymond-Hamet and Taylor, N. I. Chem. and Ind., 573 (1963).
017Larrieu, P. Thesis, Faculty of Pharmacy, University of Paris (1930).
018Lee, C. M., Trager, W.F. and Beckett, A.H. Tetrahedron, 23, 375 (1967).
019Michiels, L. and Leroux, M. Bull. Acad. Med. Belg., 5, 403, 1905.
020Phillipson, J.D., Rangsiyakul, D. and Shellard, E.J. Phytochemistry, 12, 2043 (1970).
021Raymond-Hamer. Bull. Sci. pharmacol., 46, 327 (1939).
022Raymond-Hamet. Soc. Biol. Paris, 116, 1337 (1934).
023Raymond-Hamet. Bull. Sci. Pharm. 46, 327 (1939).
024Raymond-Hamet. Ann. Pharm. France. 8, 482, (1959).
025Raymond-Hamet. Compt. rend. Acad. Sci., 235, 547 (1952).
026Raymond-Hamet and Millat, L. Bull. Sci. Pharm. 41, 533 (1934).
028Raymond-Hamet and Millat, L. Bull. Sci. Pharm. 42, 602 (1935).
029Raymond-Hamet and Millat, L. J. Pharm. Chem., 25, 391 (1937).
030Raymond-Hamer and Millat, L. Ann. Pharm. France, 47, 197 (1940).
031Ridley, H.N. Malay Plant Names (1897) cited by Hooper.
032Seaton, J. C., and Marion, L. Canad. J. Chem., 35, 1102, (1957).
033Seaton, J. C., Nair, M. D., Edwards, O. E. and Marion, L. Canad. J. Chem., 38 1935 (1960).
034Seaton, J. C., Tondeur, R. and Marion, L. Canad. J. Chem., 36, 1031 (1958).
035Shavel, J. and Zinnes, H. J. Amer. Chem. Soc., 87, 1320 (1962).
036Shellard, E.J., Beckett, A.H., Phillipson, J.D., Tantivatana, P. and Lee, C. M. Planta Medica, 15, 245 (1967).
038Shellard, E.J. and Houghton, P.J. Planta Medica, 20, 8-2 (1971).
039Shellard, E.J. and Houghton, P.J. Planta Medica, 21, 16 (1972).
040Shellard, E.J. and Houghton, P.J. Planta Medica, 21, 263 (1972).
041Shellard. E.J. and Houghton, P.J. Planta Medica, 21, 382 (1972).
042Shellard, E.J. and Houghton, P.J. Planta Medica, 22, 97 (1972).
043Shellard, E.J. and Houghton, P.J. Planta Medica, 24, 13 (1973).
044Shellard, E.J. and Houghton, P.J. Planta Medica, 24, 341 (1973).
045Shellard, E.J. and Houghton, P.J. Planta Medica, in press (1974).
046Shellard, E.J. and Phillipson, J.D. Planta Medica, 12, 27 (1964).
047Shellard, E.J. and Phillipson, J.D. Planta Medica, 12, 160 (1964).
048Shellard, E.J. and Phillipson, J.D. Tetrahedron Letters, No. 11, 1113 (1966).
049Shellard, E.J., Phillipson, J.D. and Gupta, D. Planta Medica, 16, 20 (1968).
050Shellard, E.J., Phillipson, J.D. and Gupta, D. Planta Medica, 16, 436 (1968).
051Shellard, E.J., Phillipson, J. D. and Gupta, D. Planta Medica, 17, 51 (1969).
052Shellard, E.J., Phillipson, J. D. and Gupta, D. Planta Medica, 17, 146 (1969).
053Shellard, E.J., Phillipson, J. D. And Sarpong, K. Phytochemistry, 10, 2505 (1971).
054Shellard, E.J. and Rungsiyakul, D. Planta Medica, 23, 221 (1973).
055Shellard, E.J. and Sarpong, K. Planta Medica, 20, 167 (1971).
056Shellard, E.J. and Sarpong, K. Tetrahedron, 27, 1725 (1971).
057Shellard, E.J., Tantivatana, P. and Beckett, A.H. Planta Medica, 15, 306 (1967).
058Tantivatana, Payom, private communication (1965).
059Trager, W.F., Lee, C. M. and Beckett, A.H. Tetrahedron, 23, 365 (1967).
060Trager, W. F., Lee, C. M., Phillipson, J. D. and Beckett, A.H. Tetrahedron, 23, 1043 (1967).
061Trager, W. F., Lee, C. M., Phillipson, J. D., Haddock, R. E., Dwuma-Badu, D. and Beckett, A.H. Tetrahedron, 24, 523 (1968).
062Trager, W. F., Phillipson, J. D. and Beckett, A. H. Tetrahedron, 24, 2681 (1968).
063Wenkert, E. and Bringi, N. V. J. Amer. Chem. Soc., 81, 1474.(1955).
064Wenkert, E. and Roychoudhuri, D. K. J. Amer. Chem. Soc., 78, 6417 (1956).
065Wenkert, E. and Roychoudhuri, D. K. J. Amer. Chem. Soc., 79, 1519 (1957).
066Zacharias, D.E., Rosenstein, R. D. and Jeffrey, E. A. Acta Cryst., 18, 1039 (1965).



